Genotype-by-Sequencing (GBS) is reduced representation of a genome, which utilizes restriction enzymes (e.g. ApeKI) and NextGen sequencing to identify biallelic markers and presence/absence markers. In this post, my attempt is to consisely present the GBS SNP calling process in 7 steps using the TASSEL GBSv2 pipeline. Pleae note:, Buckler et al. provides descriptive documentation on this SNP calling at this Link
Download Sample Input Files at this LINK. Please remember sample file names differs from the one mentioned in the tutorial
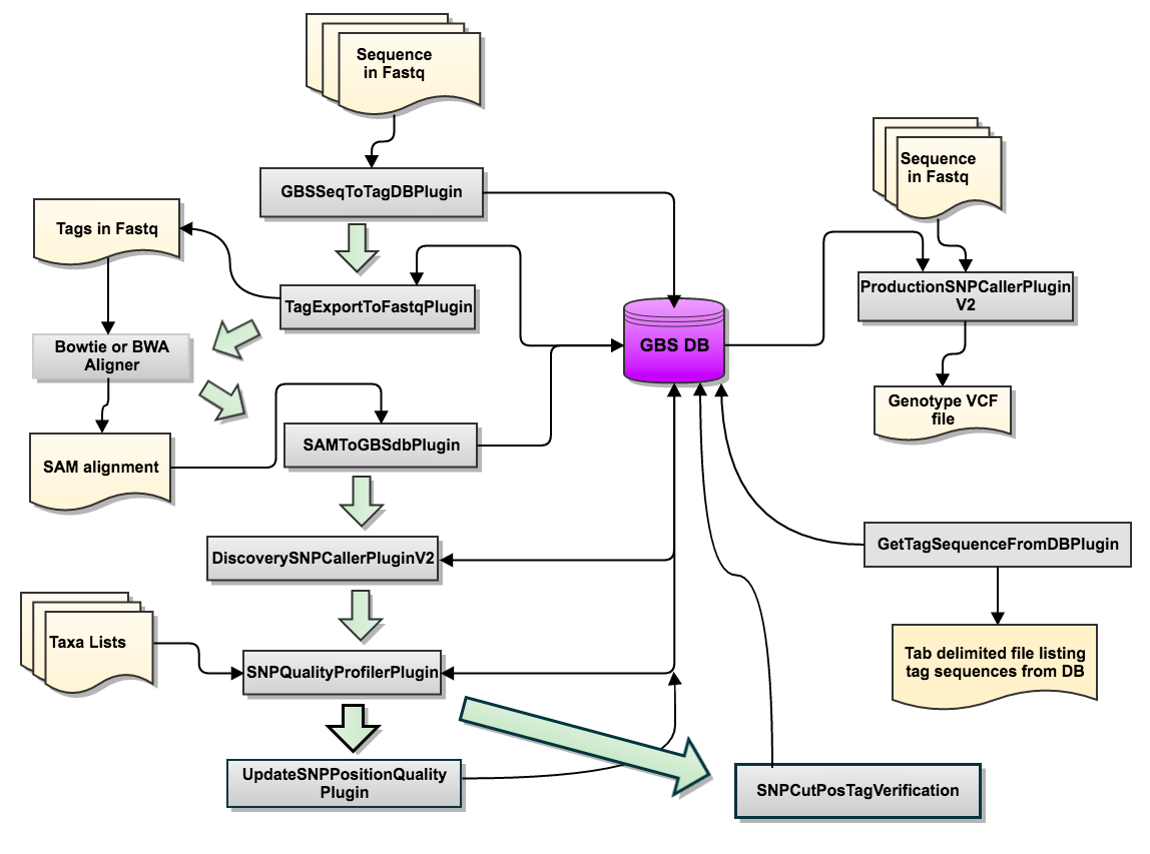
Flowchart of the GBSv2 SNP calling pipeline
Step 1. Preparing files and creating folders
To get started, create four folders named: fastq, key, output, referenceGenome, using the command below:
$ mkdir fastq key output referenceGenome
1.1 Place the GBS sequencing files (.fastq.gz) files in the fastq folder. Please remember the file names has to be in this fromat flowcell_lane_fastq.txt.gz If your fastq files does not have .fastq.txt.gzextenion, then pleae re-name them.
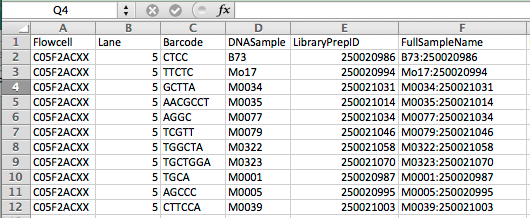
1.2 Prepare the Key file with headers and information shown below figure, and place the file in the key folder.
1.3 Download the reference genome file and place it in the referenceGenome folder.
Step 2. GBSSeqToTagDBPlugin
In this step, GBSSeqToTagDBPlugin identifies tags and the taxa from the fastq files and store in the local database.
Command:
$ /programs/tassel-5-standalone_20180419/run_pipeline.pl -fork1 -GBSSeqToTagDBPlugin -e ApeKI -i fastq/ -db output/GBSV2.db -k key/keyFile_160_271.txt -kmerLength 64 -minKmerL 20 -mnQS 20 -mxKmerNum 100000000 -endPlugin -runfork1
In the above command, ApeKI = enzyme used in the library preparation; GBSV2.db = the name of the local database.
Step 3. TagExportToFastqPlugin
In this step, TagExportToFastqPlugin is used to retrieve the distinct tags stored in the GBSV2.db database, and reformmated to the fastq tags, which can be read by the Bowtie2 aligner program. The output is a .sam file.
Command:
$ /programs/tassel-5-standalone_20180419/run_pipeline.pl -fork1 -TagExportToFastqPlugin -db output/GBSV2.db -o output/tagsForAlign.fa.gz -c 1 -endPlugin -runfork1
Step 4. Run Alignment Program(s)
4.1 Run Bowtie2 software to create an index from the reference genome.
Command:
$ bowtie2-build referenceGenome/Noirv2.upper.fa PN40024v2
PN40024v2 is the base name of the index files to write.
4.2 After the indexes have been created, the alignment command can be run. Command:
$ bowtie2 -p 15 --very-sensitive -x referenceGenome/PN40024v2/PN40024v2 -U output/tagsForAlign.fa.gz -S tagsForAlignFullvs.sam
Below is the example of the alignment statistics:
595364 reads; of these:
595364 (100.00%) were unpaired; of these:
81305 (13.66%) aligned 0 times
340723 (57.23%) aligned exactly 1 time
173336 (29.11%) aligned >1 times
86.34% overall alignment rate
Step 5. SAMToGBSdbPlugin
In this step, SAMToGBSdbPlugin reads the SAM file (output file from Bowtie alignment step) to identify the potential positions of the GBS tags against the reference genome.
Command:
$ /programs/tassel-5-standalone_20180419/run_pipeline.pl -fork1 -SAMToGBSdbPlugin -i tagsForAlignFullvs.sam -db output/GBSV2.db -aProp 0.0 -aLen 0 -endPlugin -runfork1
Below is the example output:
Total number of cut sites: 250131
Number of cut sites with 1 tag: 130620
Number of cut sites with 2 tags: 55554
Number of cut sites with 3 tags: 30647
Number of cut sites with more than 3 tags: 33310
[pool-1-thread-1] INFO net.maizegenetics.analysis.gbs.v2.SAMToGBSdbPlugin - Finished reading SAM file and adding tags to DB.
Total number of tags mapped: 514059 (total mappings 514059)
Tags not mapped: 81305
Tags dropped due to minimum mapq value: 0
Step 6. DiscoverySNPCallerPluginV2
In this step, DiscoverySNPCallerPluginV2 takes the files from the GBSV2.db database as an input and identifies SNPs from aligned tags.
Command:
$ /programs/tassel-5-standalone_20180419/run_pipeline.pl -fork1 -DiscoverySNPCallerPluginV2 -db output/GBSV2.db -sC "Noirv2.chr1" -eC "Noirv2.chr19" -mnLCov 0.1 -deleteOldData true -endPlugin -runfork1
Note: sC start chromsome and eC end chromosme. It is important to know how the chromosomes in the reference gneome are named. For example, in the above command, chromosome 1 is named as “Noirv2.chr1”.
Step 7. ProductionSNPCallerPluginV2
In this step, ProductionSNPCallerPluginV2 converts the fastq and keyfile to genotypes, then its is added to a VCF file (default).
Command:
$ /programs/tassel-5-standalone_20180419/run_pipeline.pl -fork1 -ProductionSNPCallerPluginV2 -db output/GBSV2.db -e ApeKI -i fastq/ -k key/keyFile_160_271.txt -kmerLength 64 -o 160_271_Londo_041919.vcf -endPlugin -runfork1
The output VCF file can be opened directly on GUI version of the TASSEL software.
--- End of Tutorial ---
Thank you for reading this tutorial. I really hope these steps will get you started in GBS snp calling in TASSEL. If you have any questions or comments, please let comment below or send me an email.
Happy SNP calling !
Bibliography
Glaubitz, J. C., Casstevens, T. M., Lu, F., Harriman, J., Elshire, R. J., Sun, Q., & Buckler, E. S. (2014). TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PloS one, 9(2), e90346.