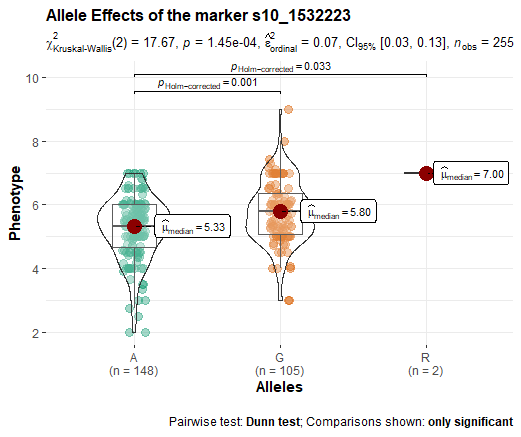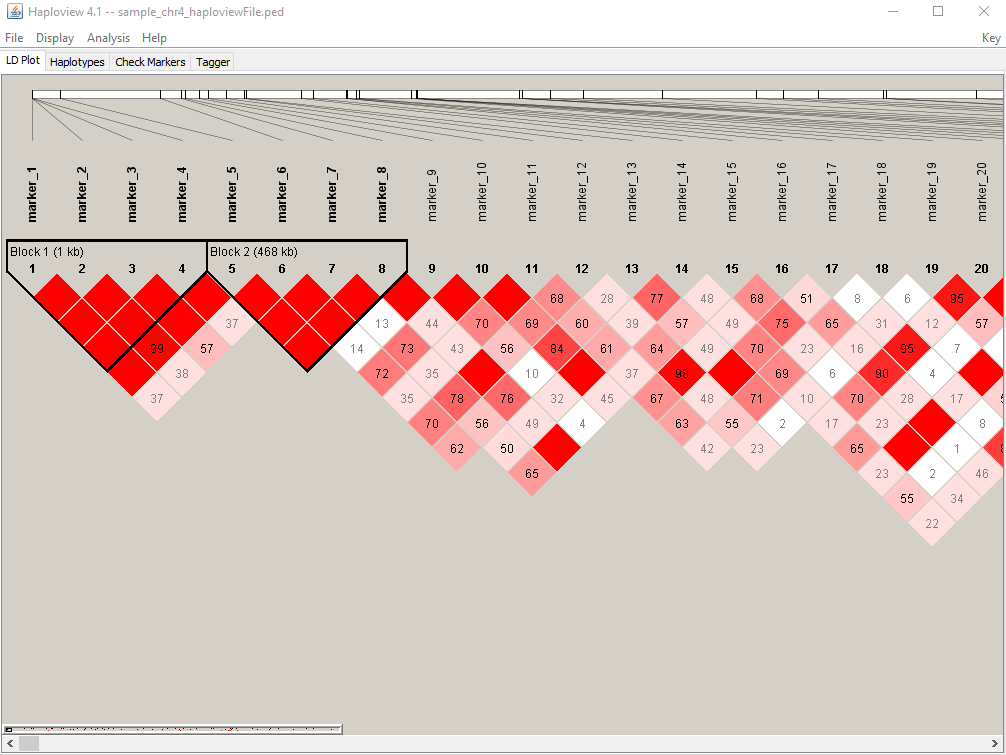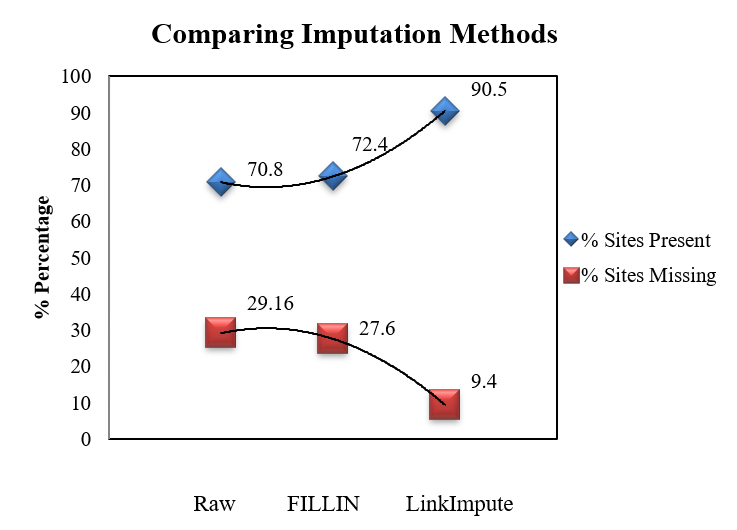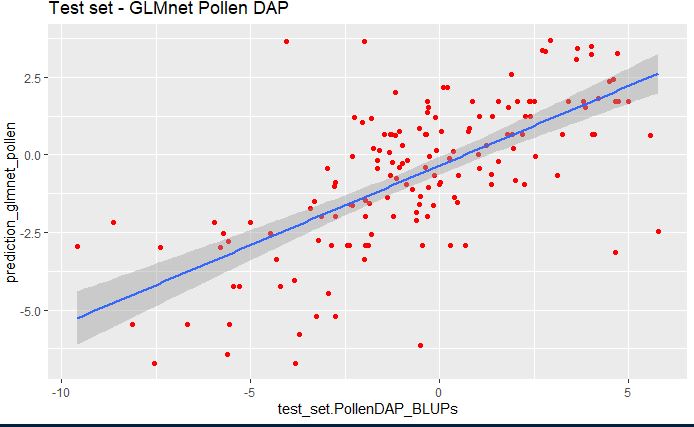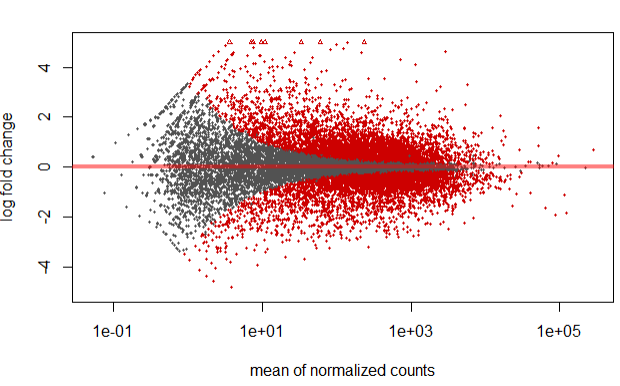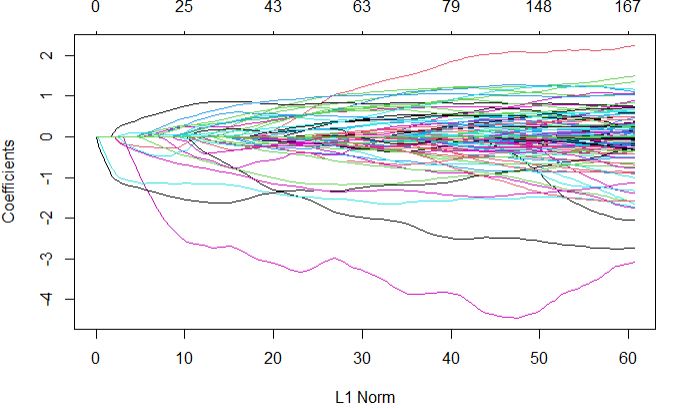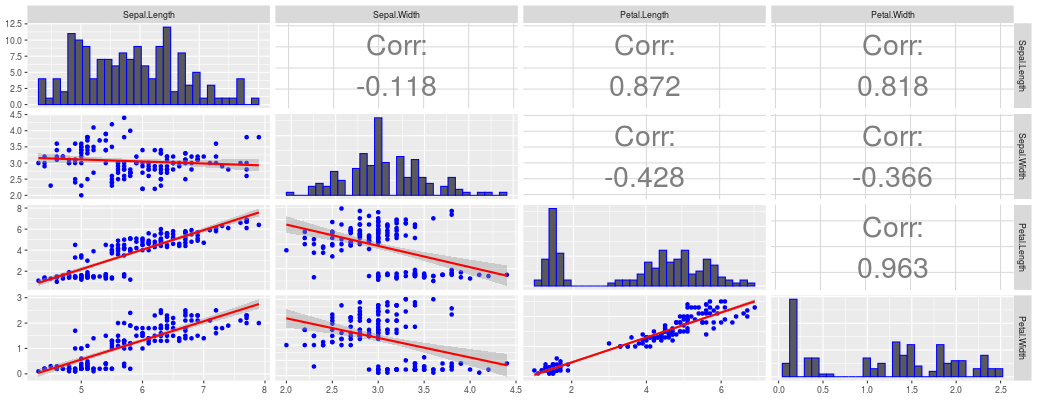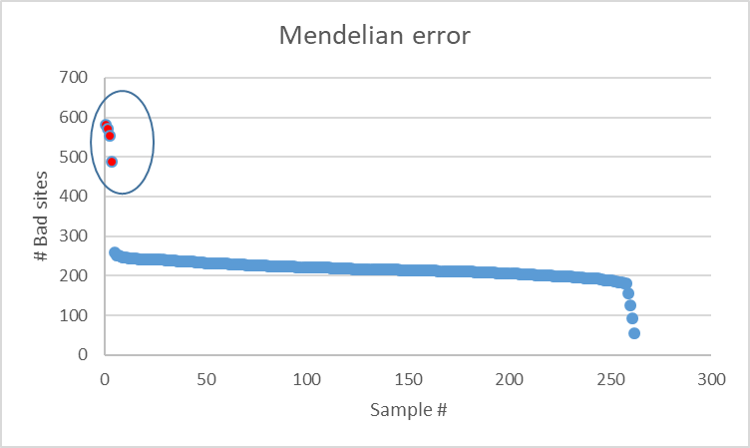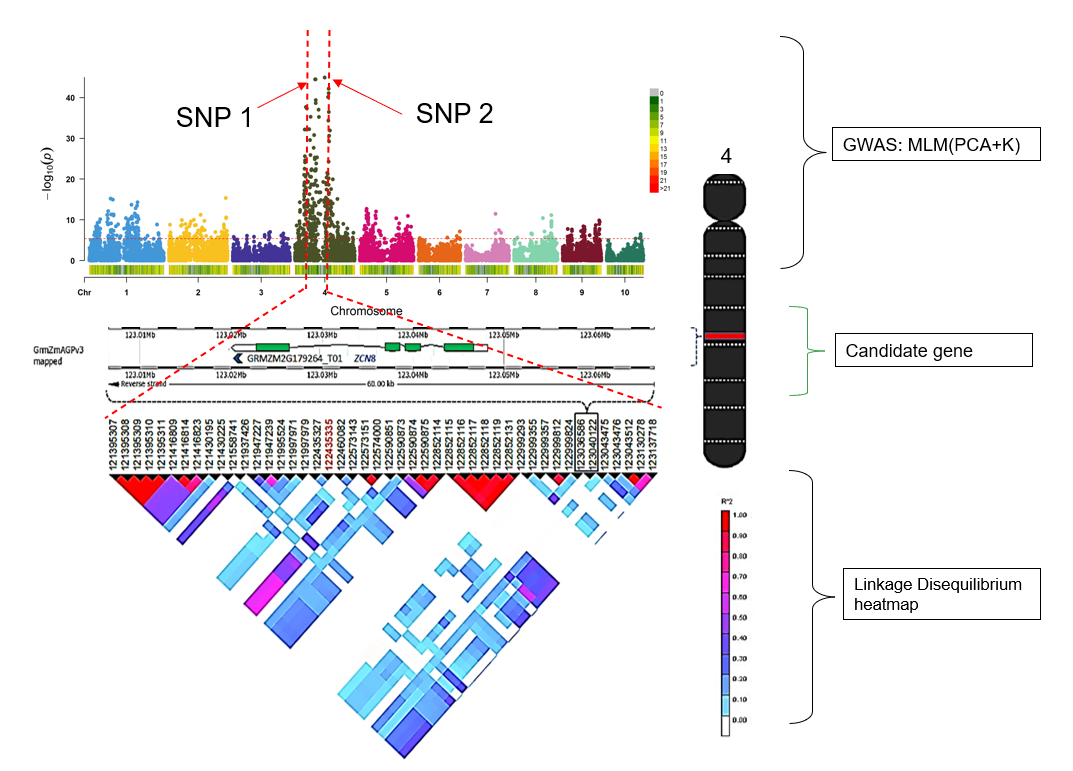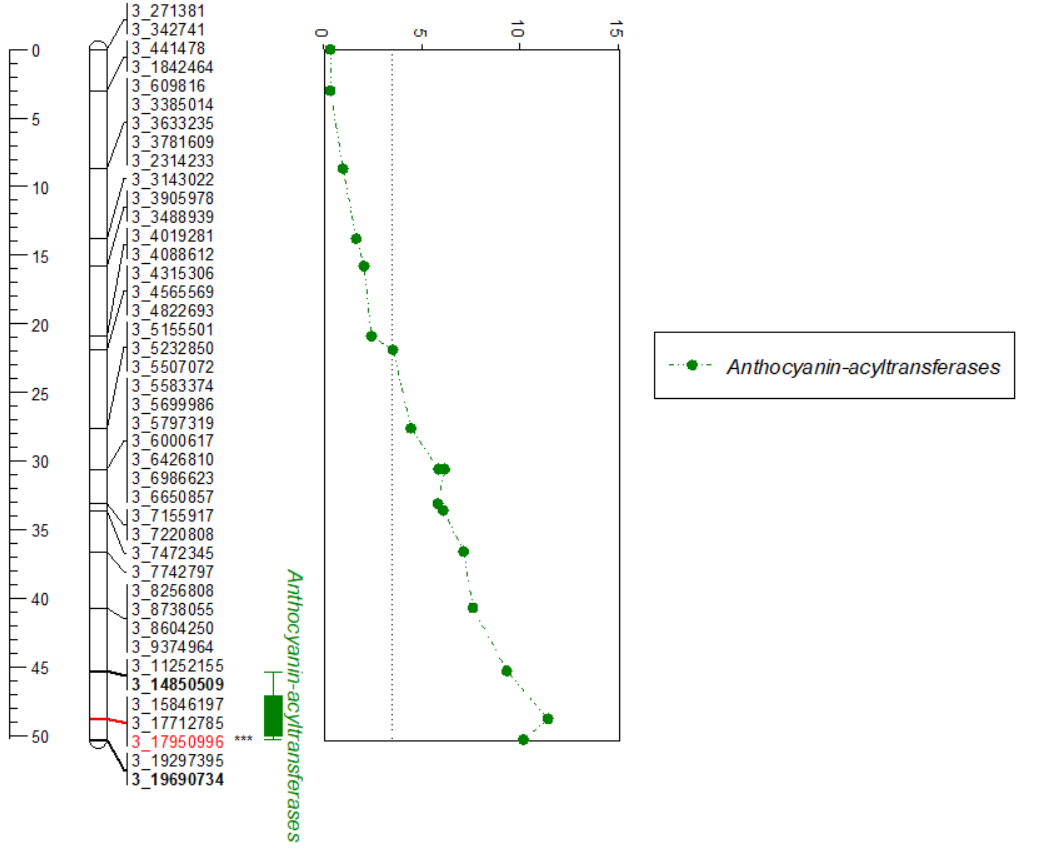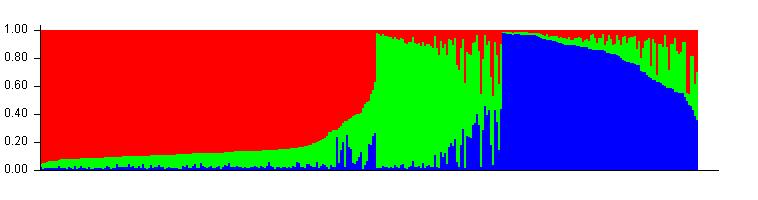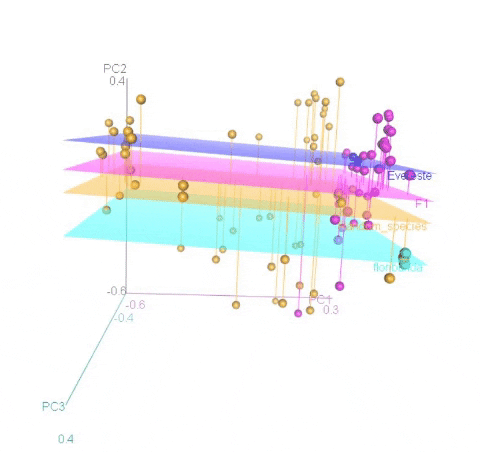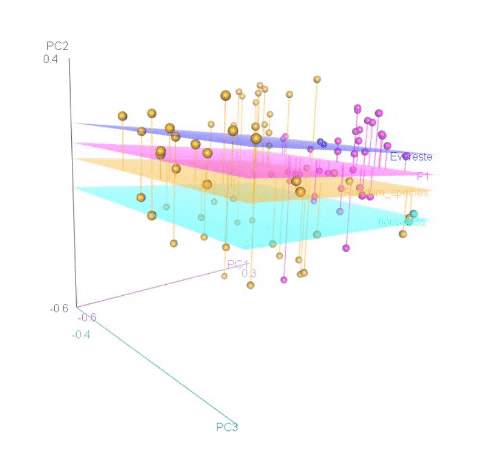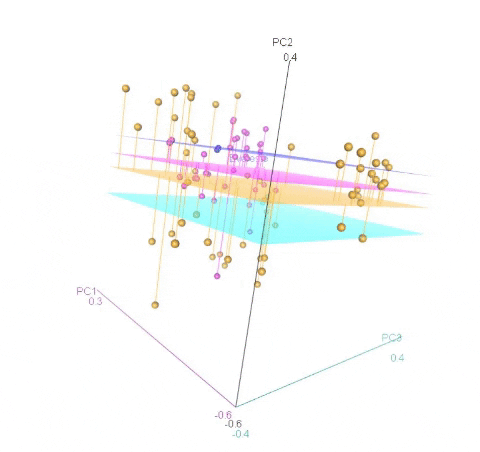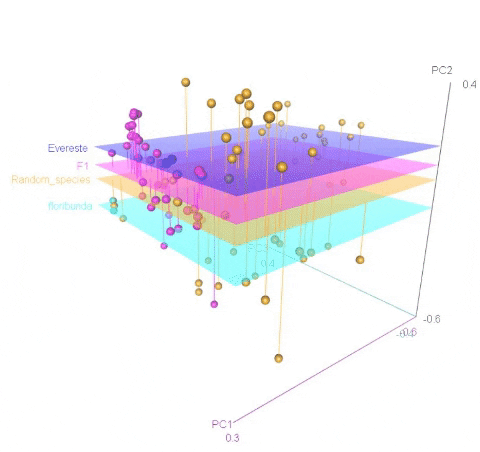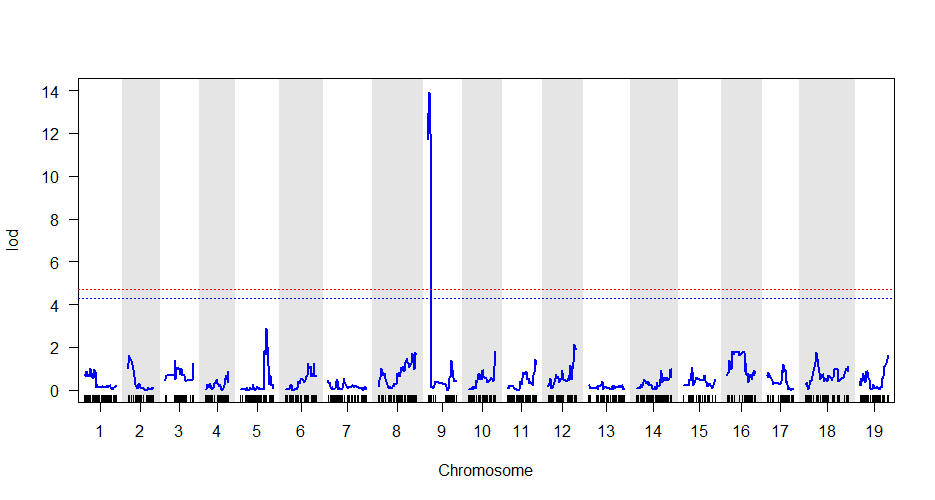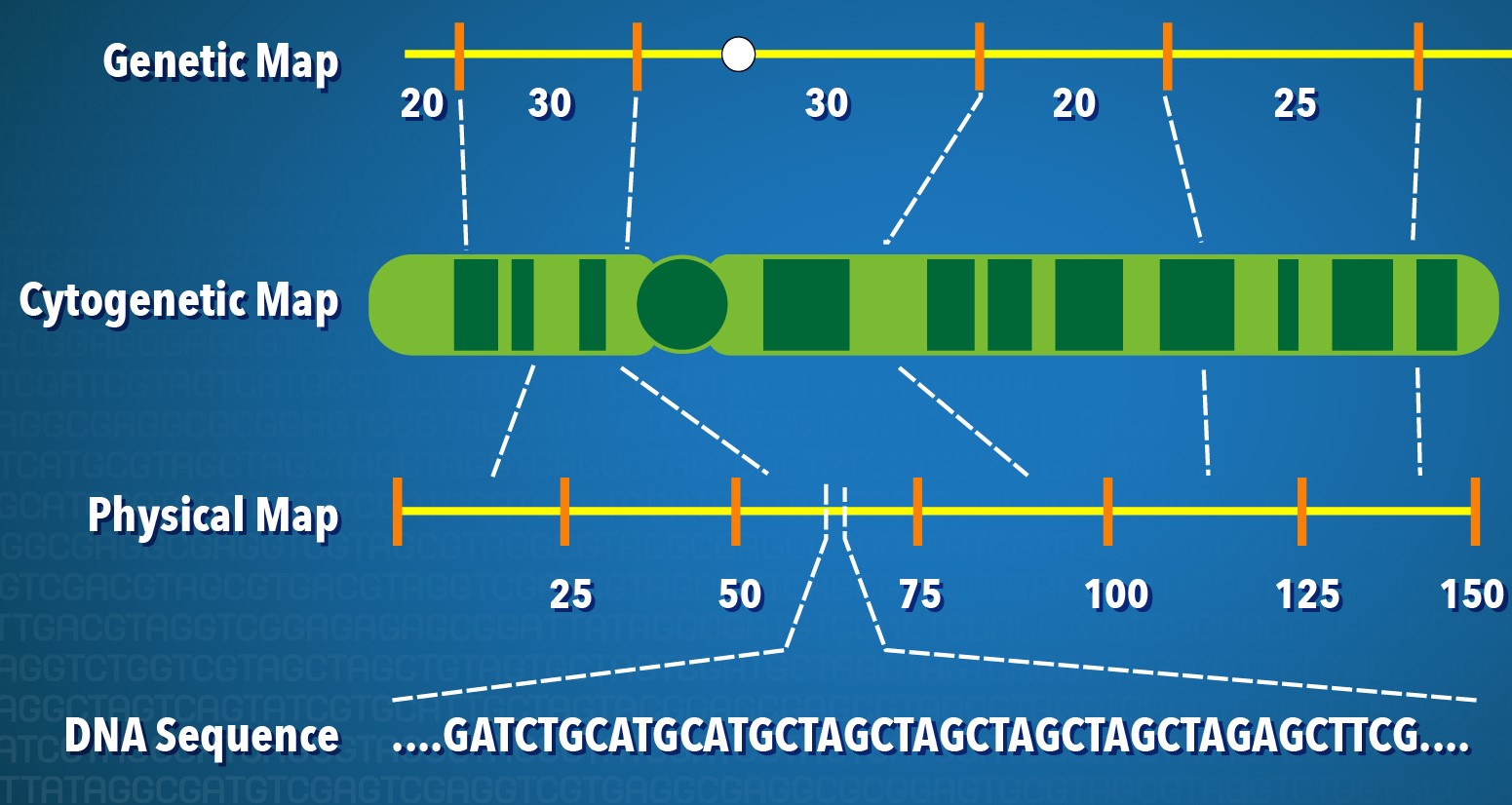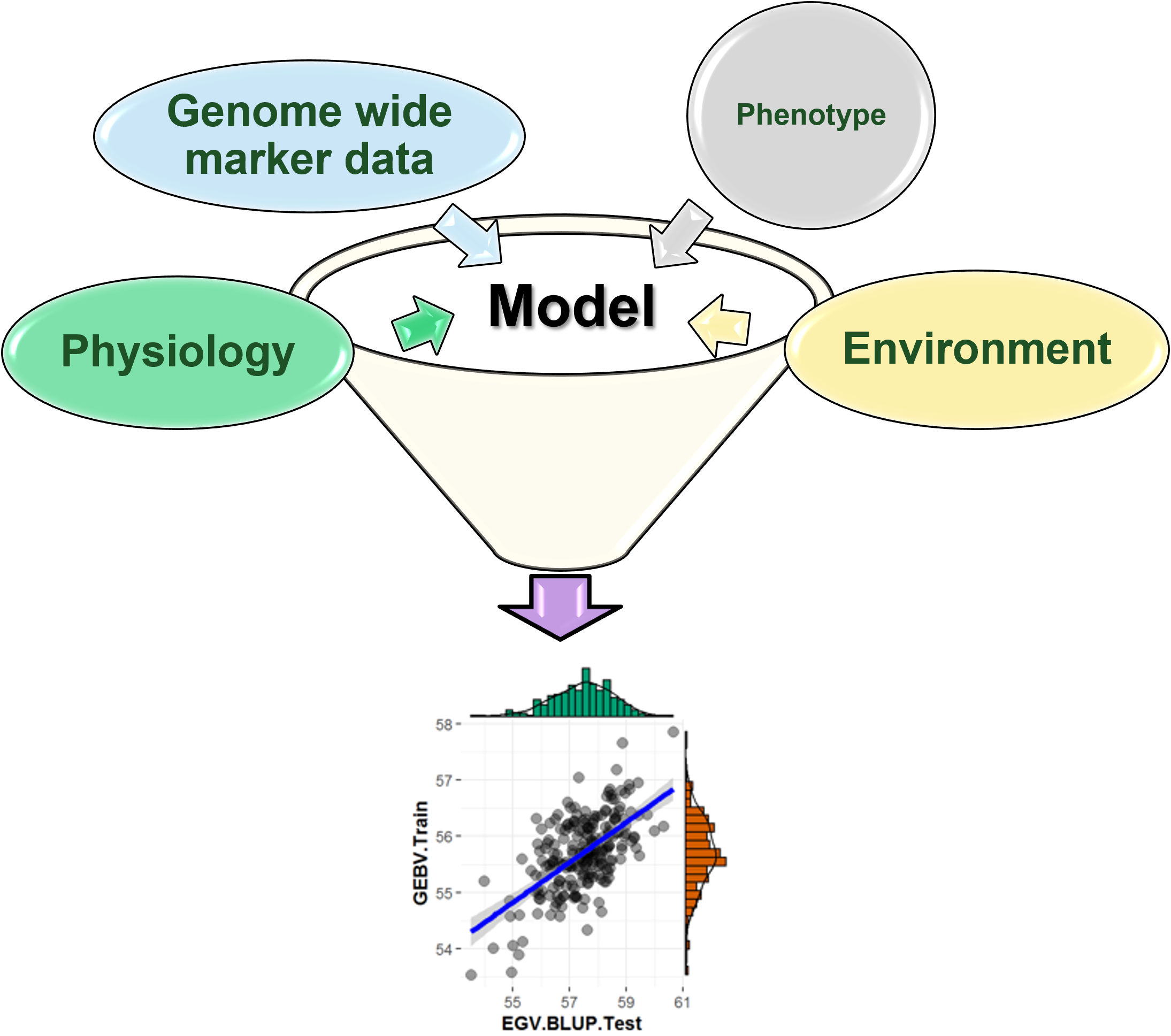
Meuwissen et al 2000 first proposed genomic selection to predict breeding values in animals and plants – the idea was to use DNA markers as predictors in model development to estimate marker effects to predict genomic estimated breeding values. Genomic selection (GS) is becoming a popular technique enabling breeders to...
[Read More]