High density markers do not provide any additional information, therefore, can be pruned based on the physical distances between adjacent markers and linkage disequilibrium (LD). In this tutorial, I will show how to prune markers based on their physical position in TASSEL software, and based on LD in PLINK software.
Pruning markers based on the physical distances in TASSEL
For detailed information on how to use TASSEL software, please consult user’s guide and further documentation at:
https://www.maizegenetics.net/tassel
1.1 Download and install TASSEL software
Download and install the latest version of the TASSEL software at this link:
https://www.maizegenetics.net/tassel
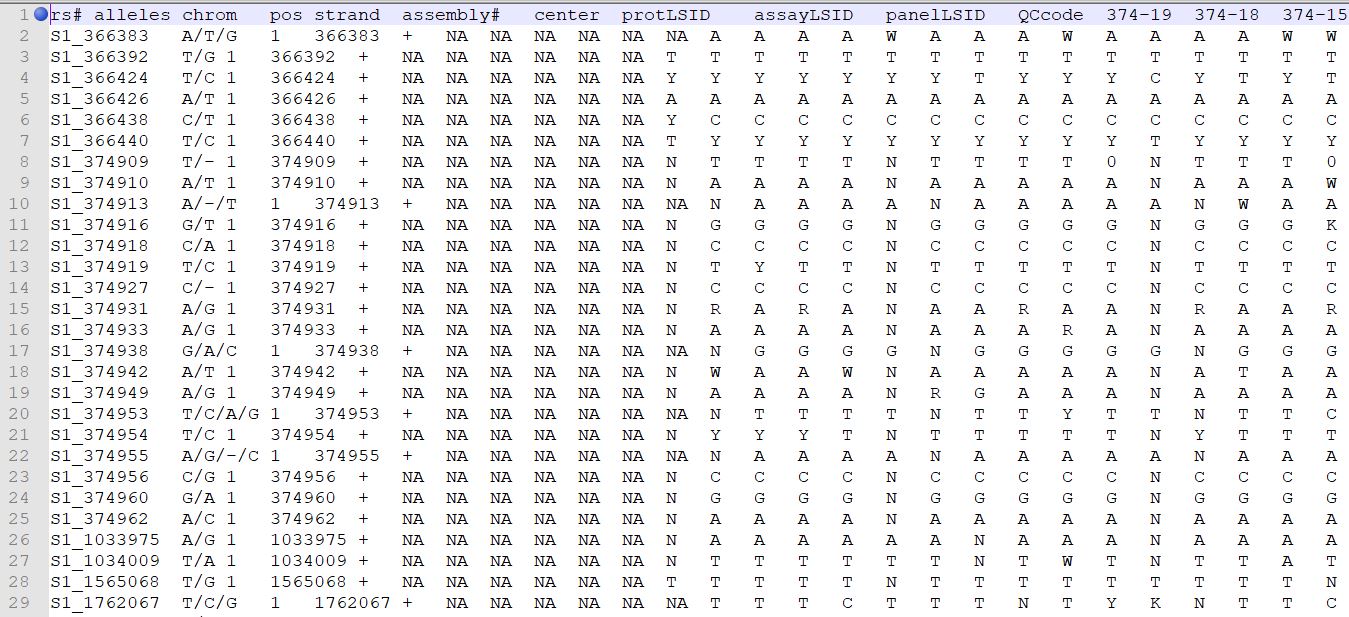
Genotype file
TASSEL allows various genotype file formats such as VCF (variant call format), .hmp.txt, and plink. In this tutorial, I am using the hmp.txt version of the genotype file. The below is the screenshot of the hmp.txt genotype file.
Step 1.2: Importing files
Import the files by following the steps shown below.
Tip! Both files can be opened at same time holding CTRL and clicking the file names.
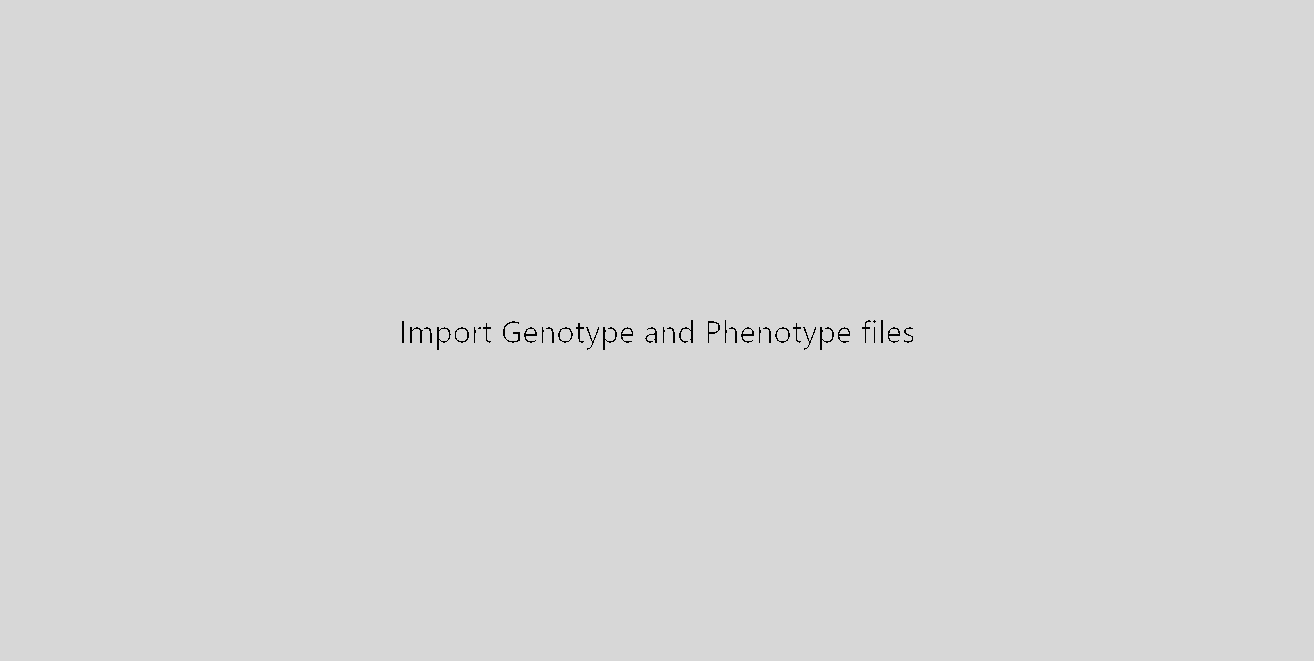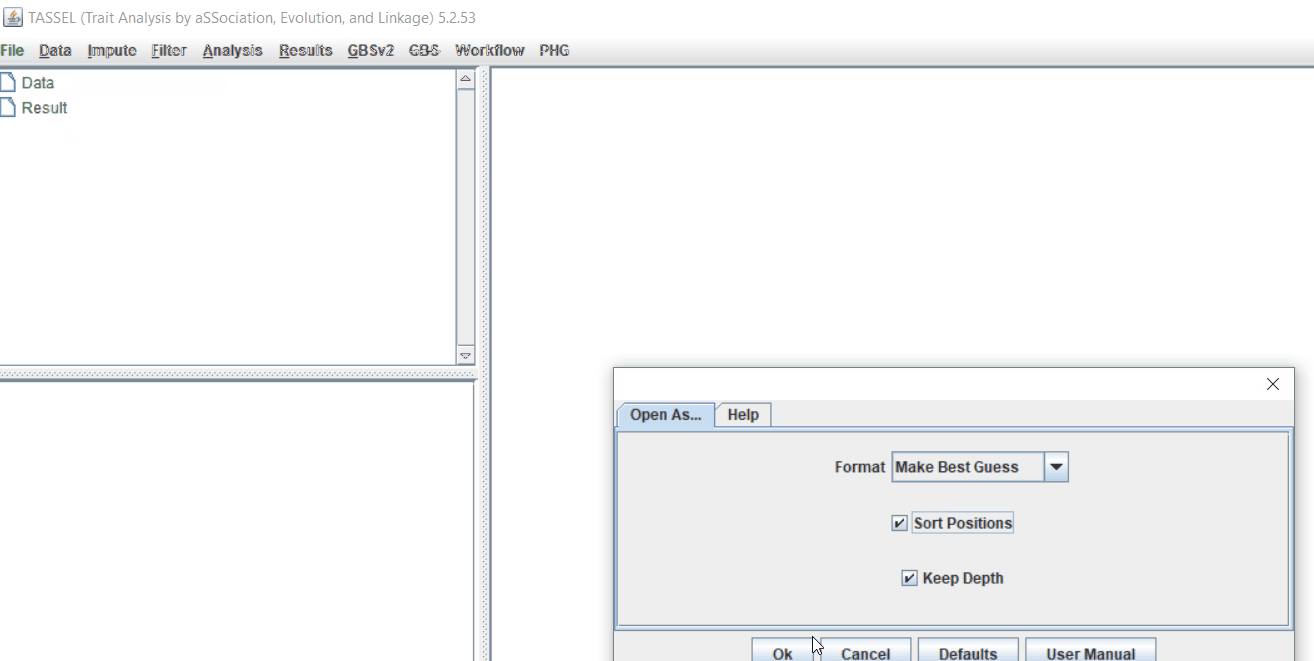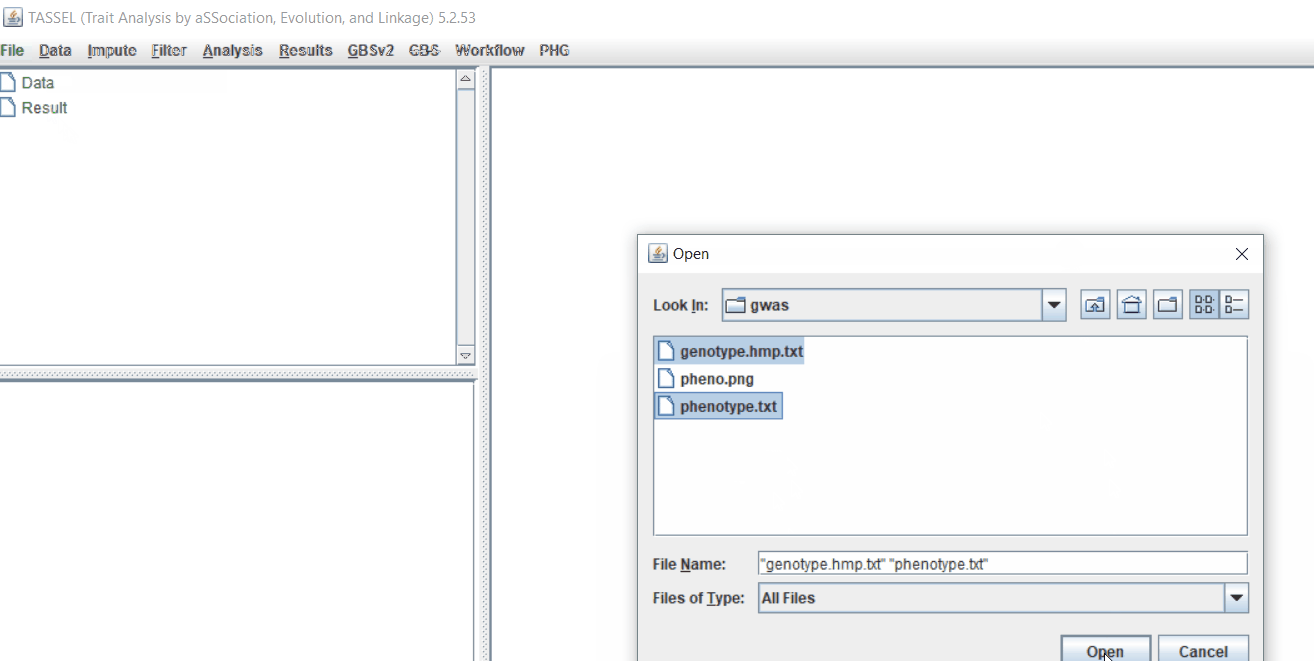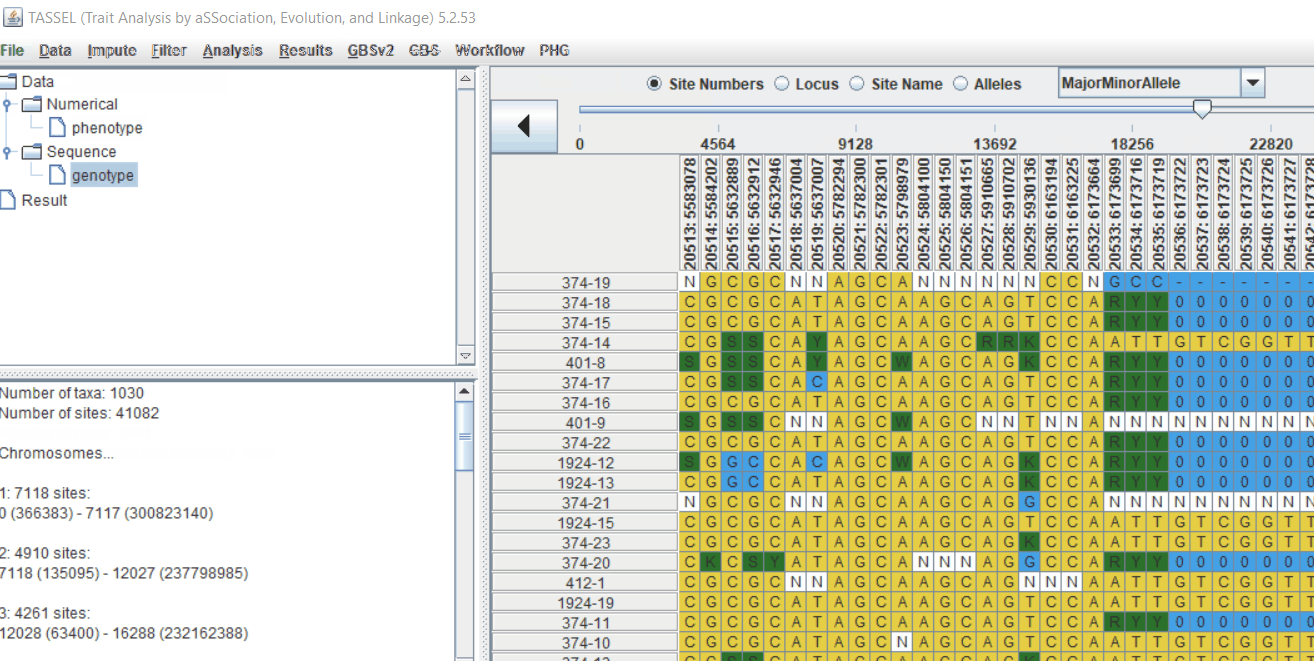
Command line pruning in TASSEL
Use below sample command line to filter :
./run_pipeline.pl -importGuess /Users/lcj34/genotyep.hmp.txt -ThinSitesByPositionPlugin
-o /Users/lcj34/thin40000.vcf -minDist 40000 –endPlugin
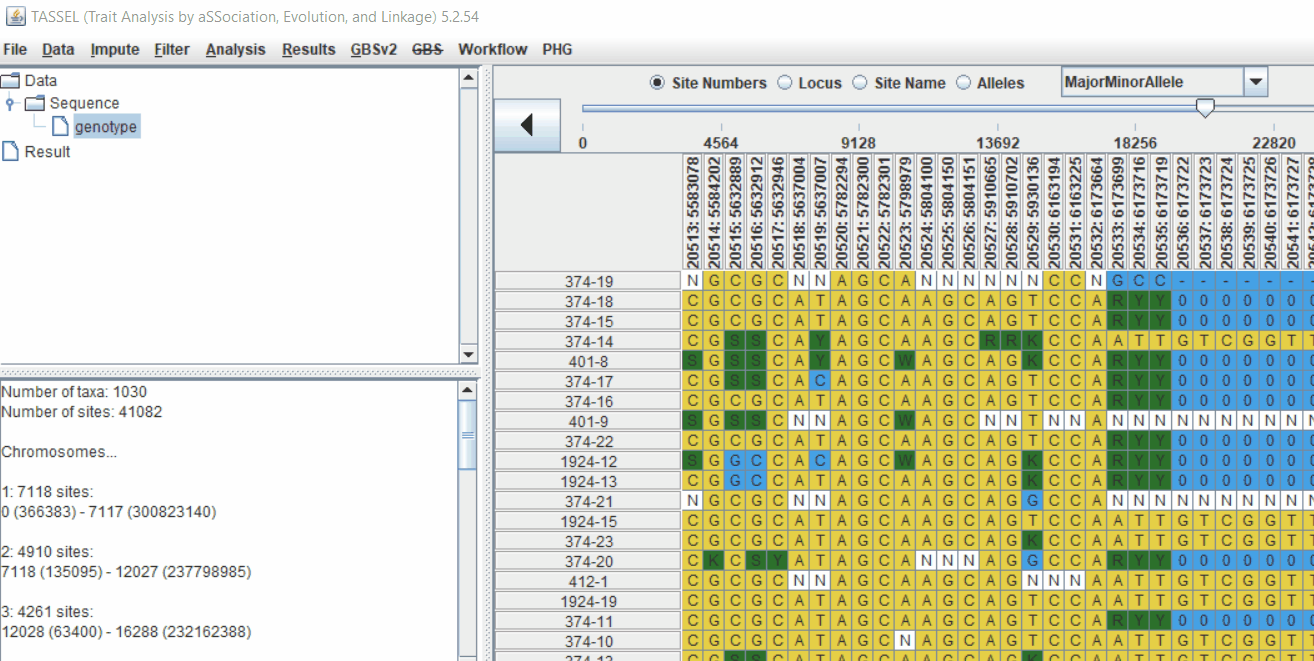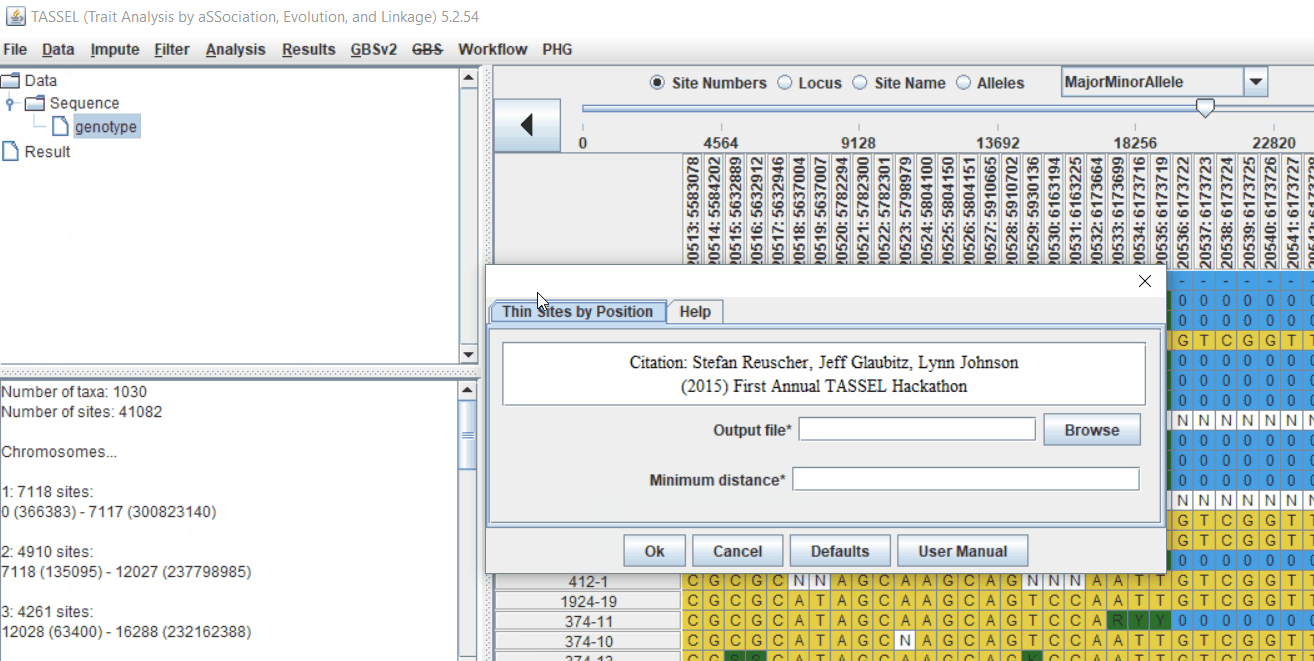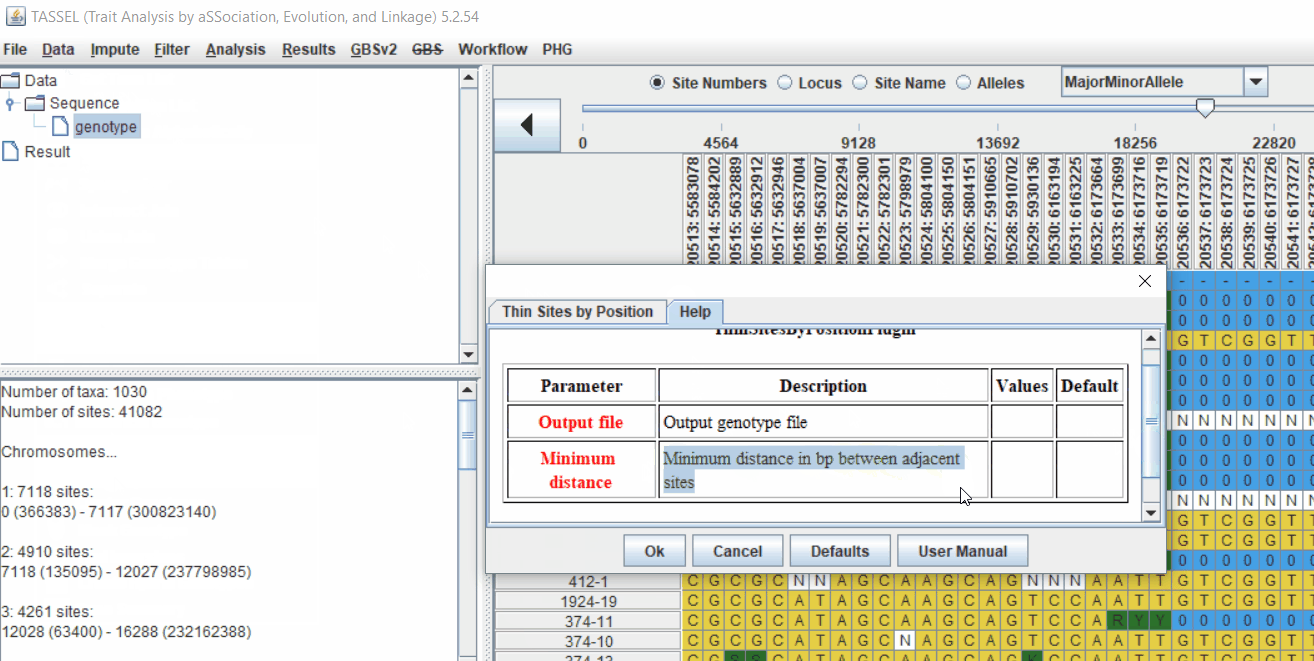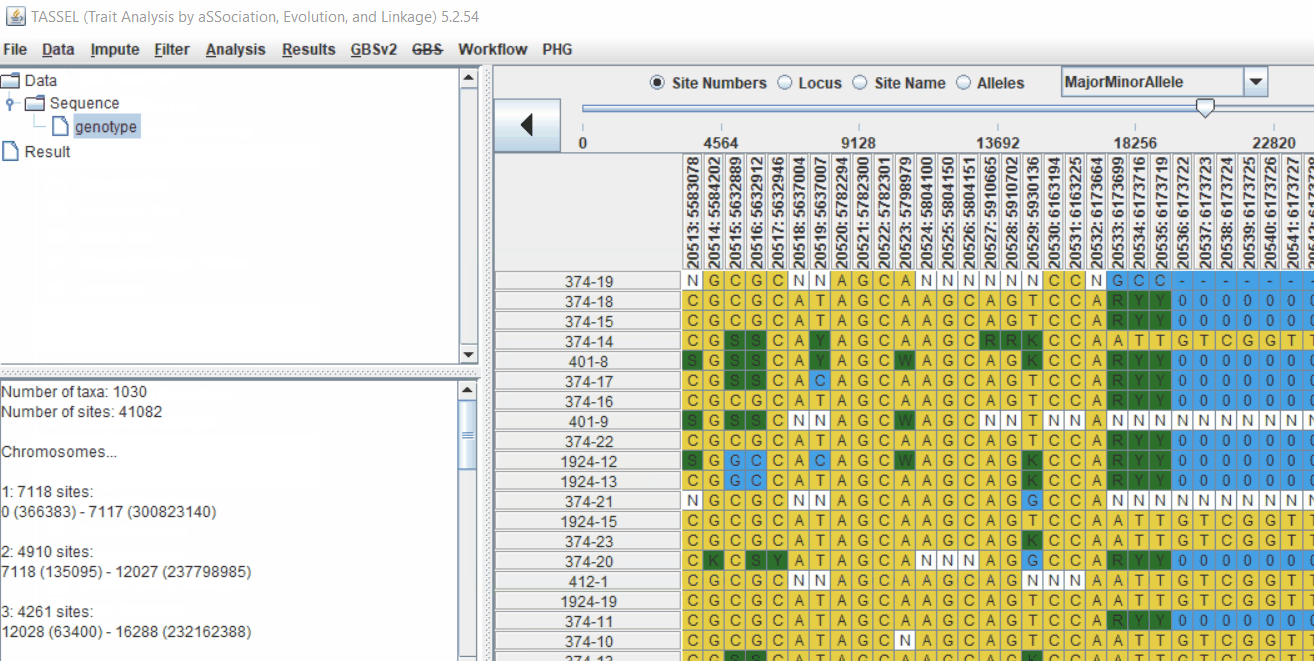
Pruning in GUI of TASSEL
Or else, in TASSEL GUI, you can use “Thin Sites by Position” plugin:
2.0 LD based pruning in PLINK software
1.1 Download and install PLINK software
Download and install the latest version of the PLINK software at this link:
http://zzz.bwh.harvard.edu/plink/download.shtml
Note: While downloading the software make sure you choose your correct platform!
2.1 Converting your genotype data into PLINK format
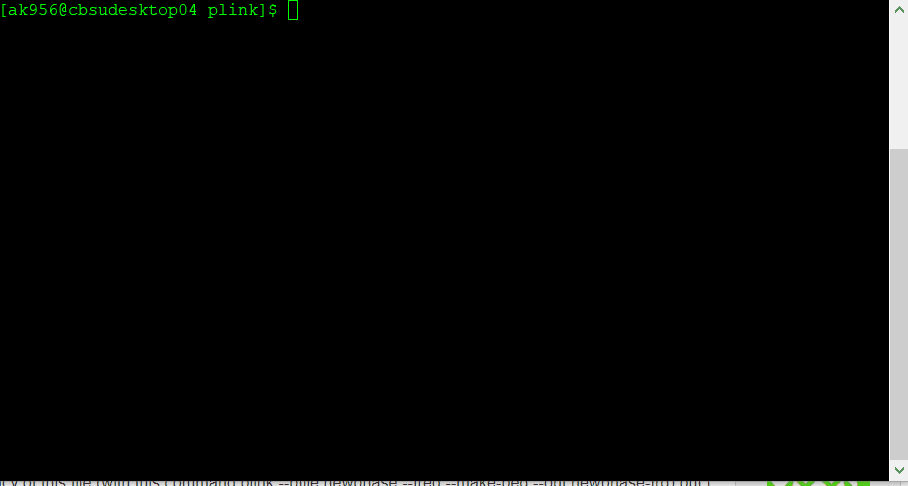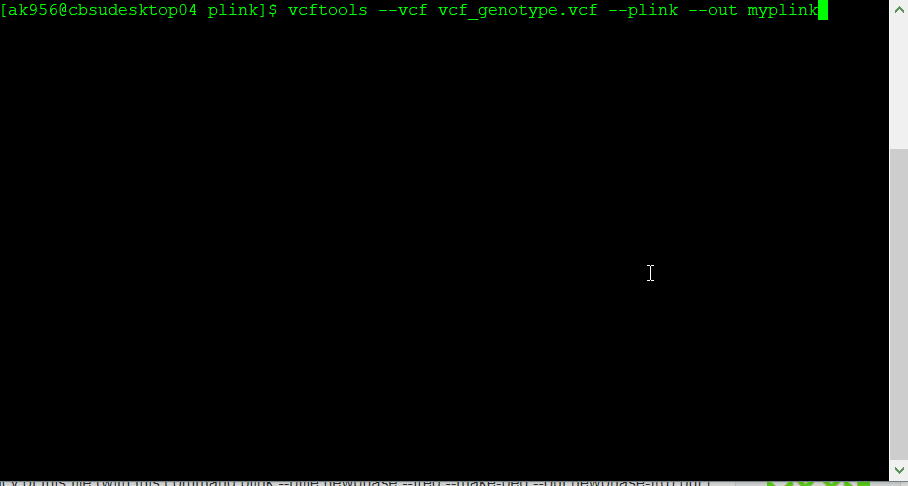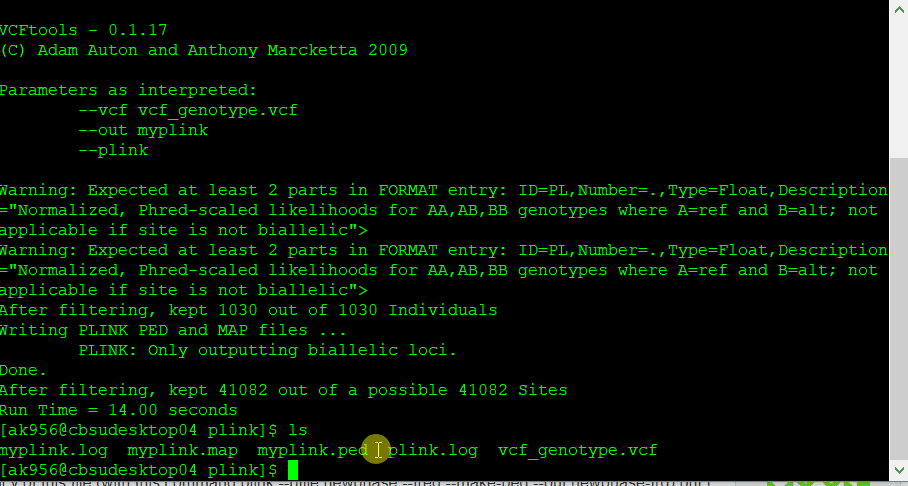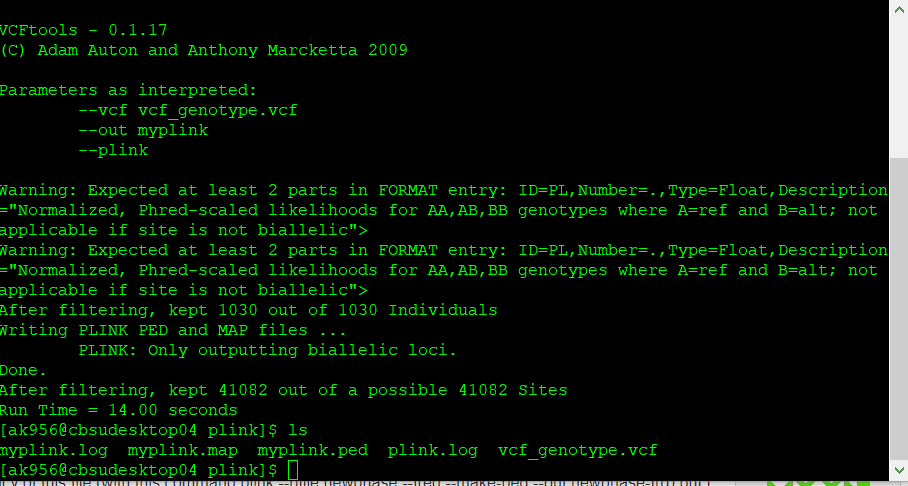
If you have a genotype data in formats such as VCF, then you will need to convert it to PLINK format in VCF tools using the command line below:
vcftools --vcf myvcf.vcf --plink --out myplink
Note: Your genotype file has to be in the VCF format in order to convert into PLINK format
2.2 Extracting markers for pruning based on LD in PLINK
PLINK has two options for LD thinning/pruning: based on variance inflation factor (by regressing a SNP on all other SNPs in the window simultaneously) and based on pairwise correlation (R2). These are the --indep and --indep-pairwise options, respectively.
Syntax for `–indep-pairwise’:
--indep-pairwise <window size>['kb'] <step size (variant ct)> <r^2 threshold>
Below is the code:
plink --noweb --file data_in --indep-pairwise 50 5 0.5 --out data_out
The command above that specifies 50 5 0.5 would
- consider a window of 50 SNPs
- calculate LD between each pair of SNPs in the window
- remove one of a pair of SNPs if the R2 is greater than 0.5
- shift the window 5 SNPs forward and repeat the procedure
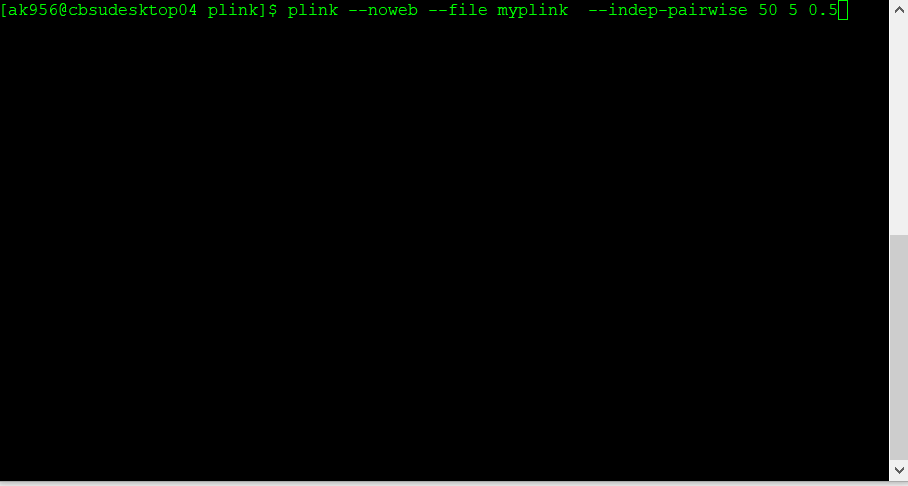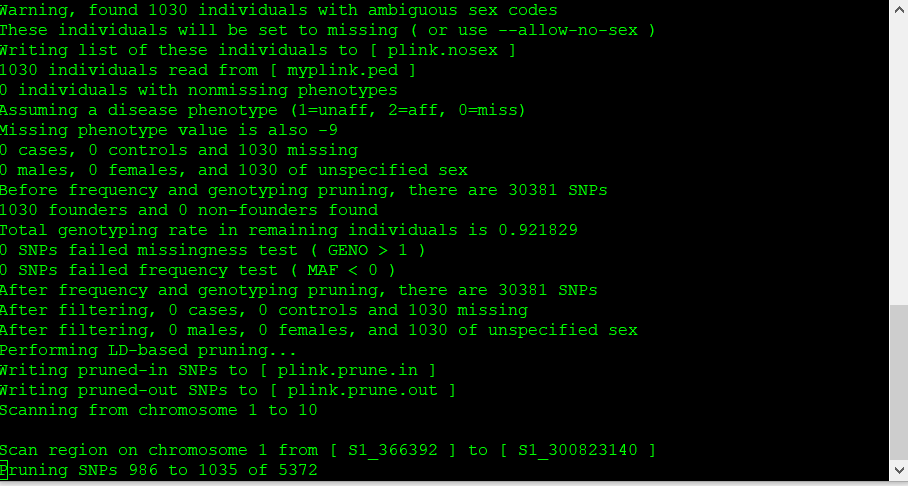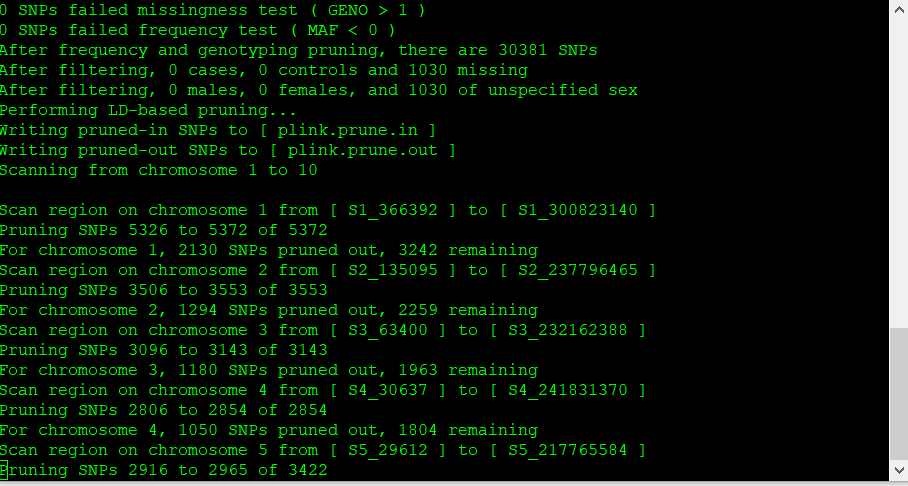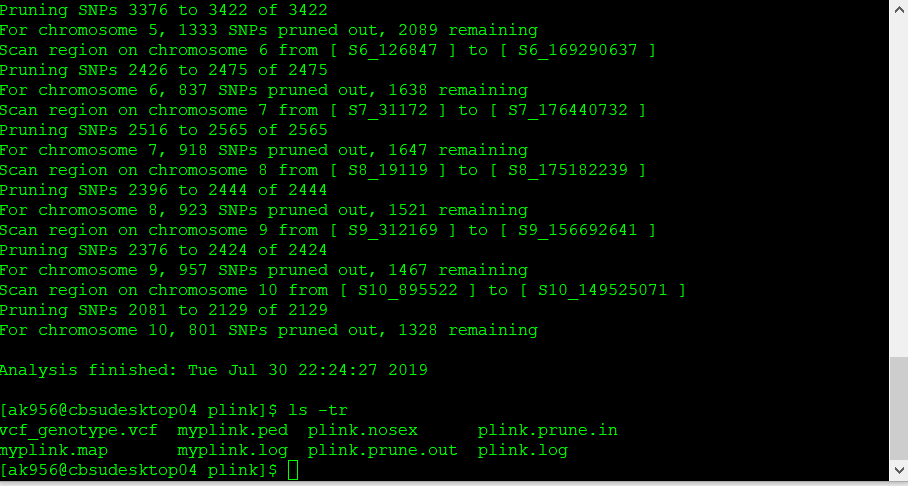
The output of the above commands creates two lists of SNPs: those that are pruned out and those that are not. See below:
plink.prune.in
plink.prune.out
2.3 Make a new, pruned file
Next, make a new, pruned file using the command below:
plink --file data --extract plink.prune.in --make-bed --out pruneddata
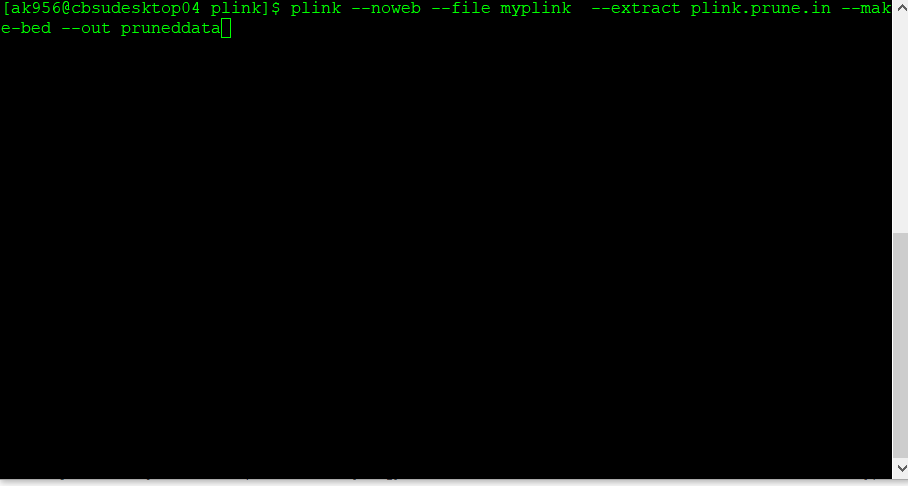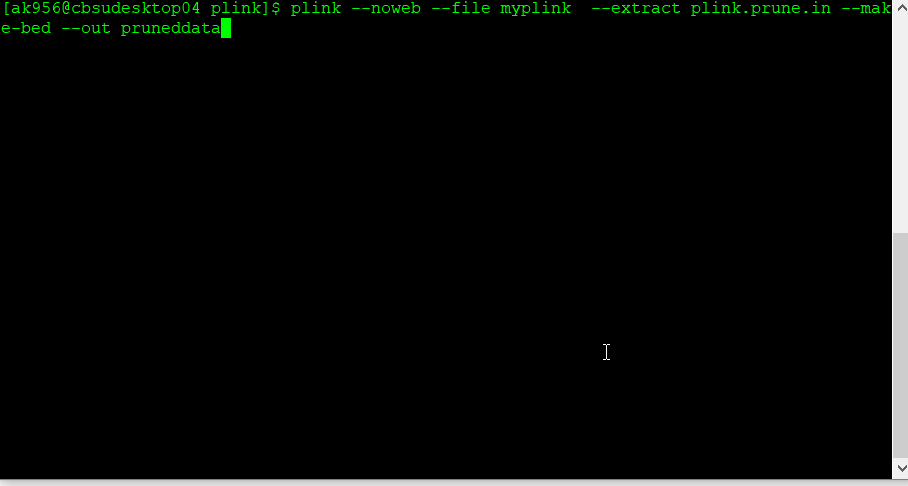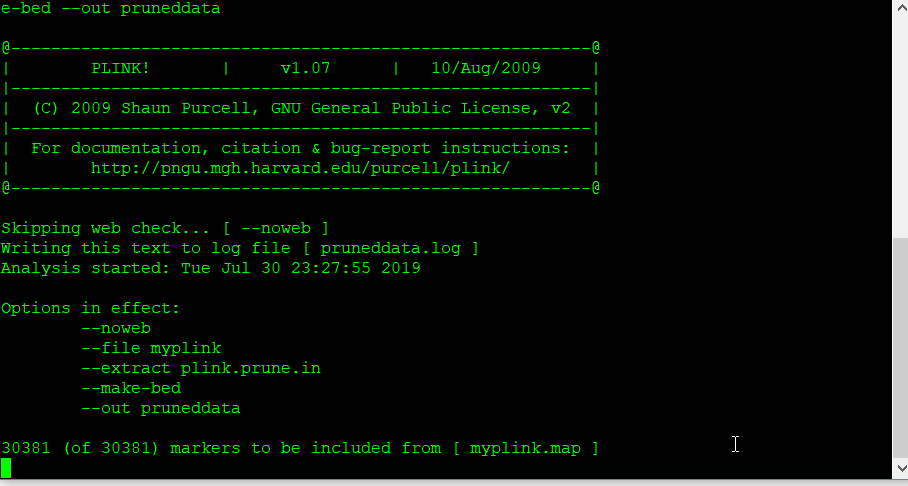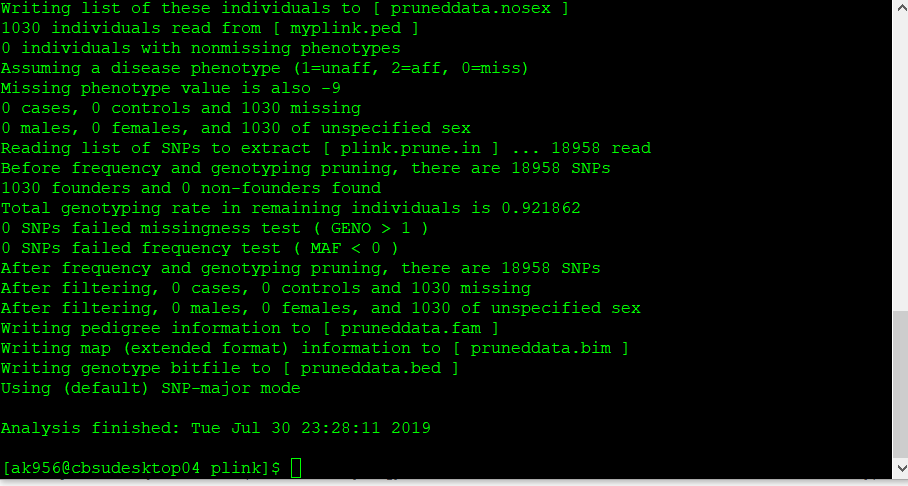
2.4 Convert the prunned output file into VCF
The pruned file can be converted back to VCF format using the command below:
plink109 --bfile pruneddata --recode vcf --out vcf_pruned
Note: please remember this parameter is only available in PLINK v 1.09
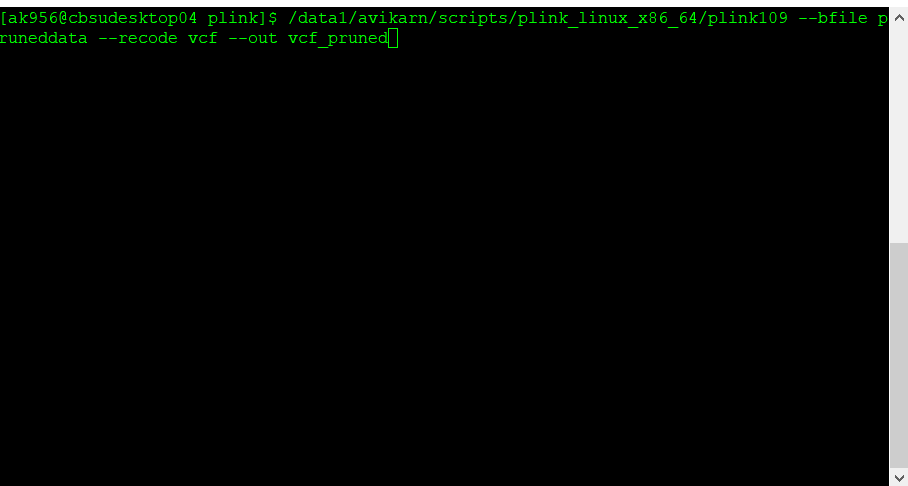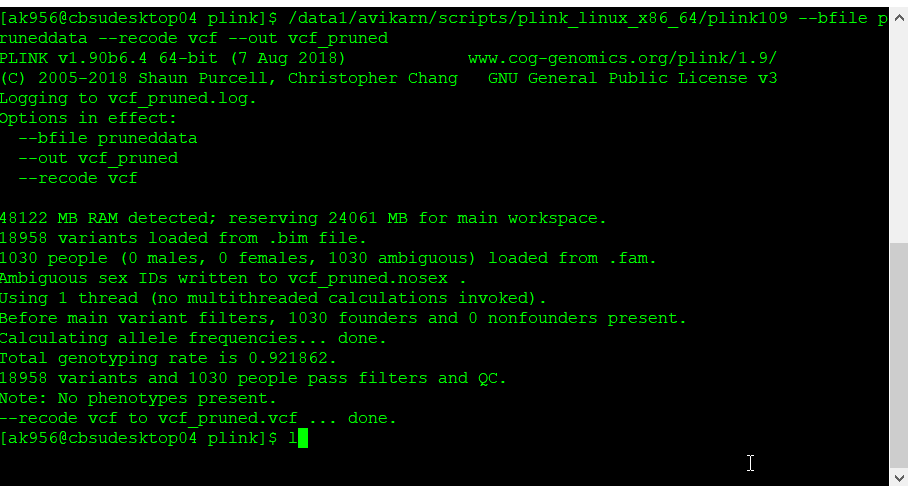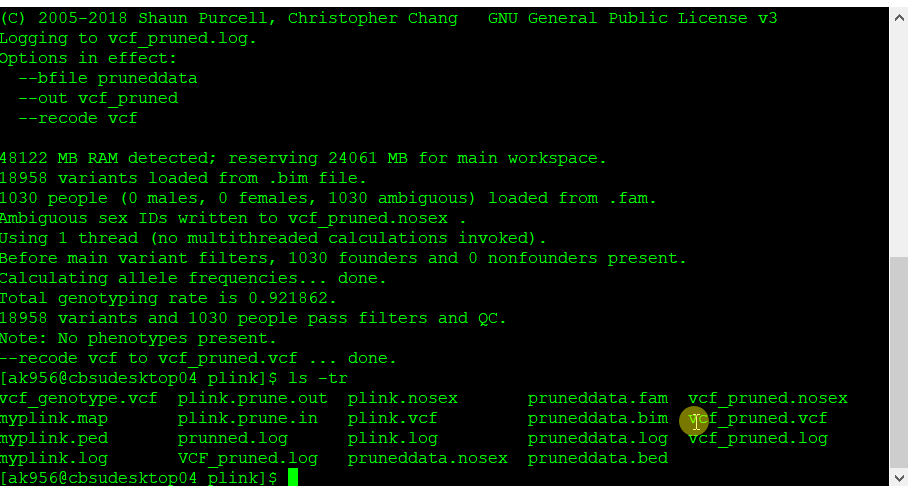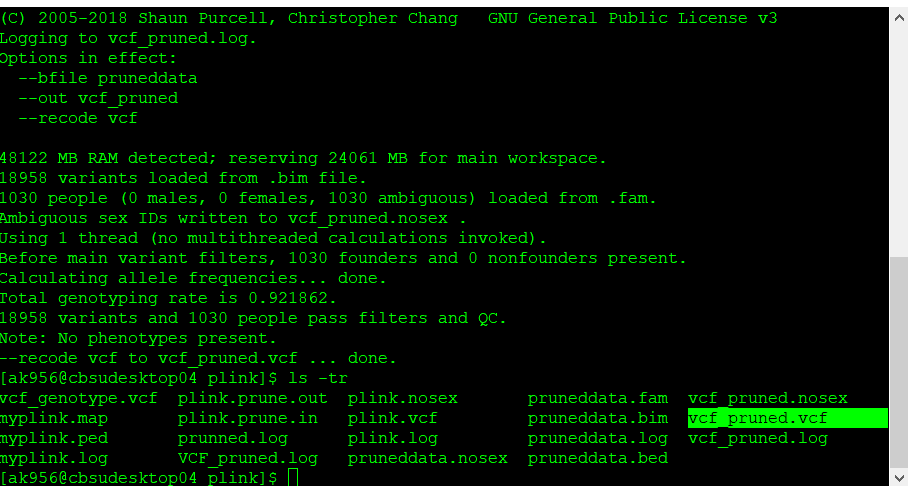
Once the final pruned data is converted to VCF file it can be viewed in TASSEL software for further analysis.
--- End of Tutorial ---
Thank you for reading this tutorial. If you have any questions or comments, please let me know in the comment section below or send me an email.
Bibliography
Package: PLINK (including version number) Author: Shaun Purcell URL: http://pngu.mgh.harvard.edu/purcell/plink/ Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ & Sham PC (2007) PLINK: a toolset for whole-genome association and population-based linkage analysis. American Journal of Human Genetics, 81.