Cluster based methods such as multidimensional scaling (MDS) and priniciple component analysis (PCA) are traditionally used in identifying samples with genotypic inconsistencies, however, it is important to identify genotypes with high mendelian inconsistencies prior to any genetic or statistical analysis. In this tutorial, I would like to share how one can quickly check samples with high mendelian inconsistencies using genotypic data and pedigree information in bcftools.
Installing bcftools
bcftools is only available for Linux operating system. For detailed information on how to install it please consult user’s guide and further documentation at below link: https://samtools.github.io/bcftools/howtos/install.html
Input data
Two files are required as:
- Genotype file in VCF format
- Pedigree file in text (.txt) version
Pedigree file format
The pedigree file should be formatted as shown below:
mother,father,child1
mother,father,child2
mother,father,child3
mother,father,child4
mother,father,child5
mother,father,child6
mother,father,child7
mother,father,child8
mother,father,child9
mother,father,child10
mother,father,child11
mother,father,child12
mother,father,child13
mother,father,child14
mother,father,child15
mother,father,child16
Important! Make sure the parent names are sperated by comma (,) without any spaces in the text file.
running the bcftools
Use below sample command line to run the program:
$ bcftools +mendelian file.vcf -T pedigree.txt -c > output
Output
The out file contains the following headers:
# [1]nOK [2]nBad [3]nSkipped [4]Trio
334518 4621 996 mother,father,child
Where,
- nOK .. number of genotypes with no missing sites and no Mendelian error
- nBad .. number of genotypes with a Mendelian error
- nSkipped .. number of genotypes at which at least one individual missing and therefore could not be considered 4
Plotting output
The output file can be opened directly in MS Excel and can be interpreted by plotting it.
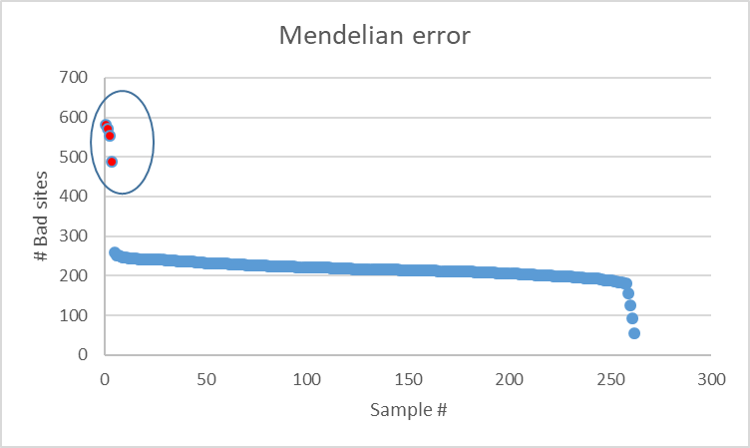
In the above plot, it can be noted that there are four samples (in red) that have the highest mendelian error in comparison to the rest of samples, which should be excluded from any further analysis.
--- End of Tutorial ---
Thank you for reading this tutorial. If you have any questions or comments, please let me know in the comment section below or send me an email.
Bibliography
*Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. and 1000 Genome Project Data Processing Subgroup (2009) The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics, 25, 2078-9. [PMID: 19505943]